Chantier 3 : l'évaluation et l'efficacité administrative pour un accès à l'innovation plus rapide
Pourquoi ?
L'innovation de rupture peine à être reconnue dans le système d'évaluation et de fixation du prix des médicaments, qui a été conçu pour accompagner les innovations progressives, non les innovations de rupture.
Or, l'innovation expérimentale, avec l'évolution de la recherche clinique issue des biotechnologies, l'immunothérapie ou encore l'essor des thérapies géniques dans les maladies rares, bouleverse ce modèle.
Le système de santé ne peut donc plus faire l'économie d'une refondation de la doctrine d'évaluation de l'innovation en santé pour s'adapter au rythme des révolutions médicales des biotechnologies et de la thérapie génique.
Il ne s'agit pas seulement de faire évoluer les critères d'évaluation pour qu'ils coïncident avec le potentiel de progrès thérapeutique : il s'agit aussi de simplifier la mise à disposition de médicaments innovants auprès des patients avant la fin de leur évaluation.
Nouvelles immunothérapies anticancéreuses par indication : état des autorisations de mise en marché et des inscriptions au remboursement aux Etats-Unis, en Europe et en France
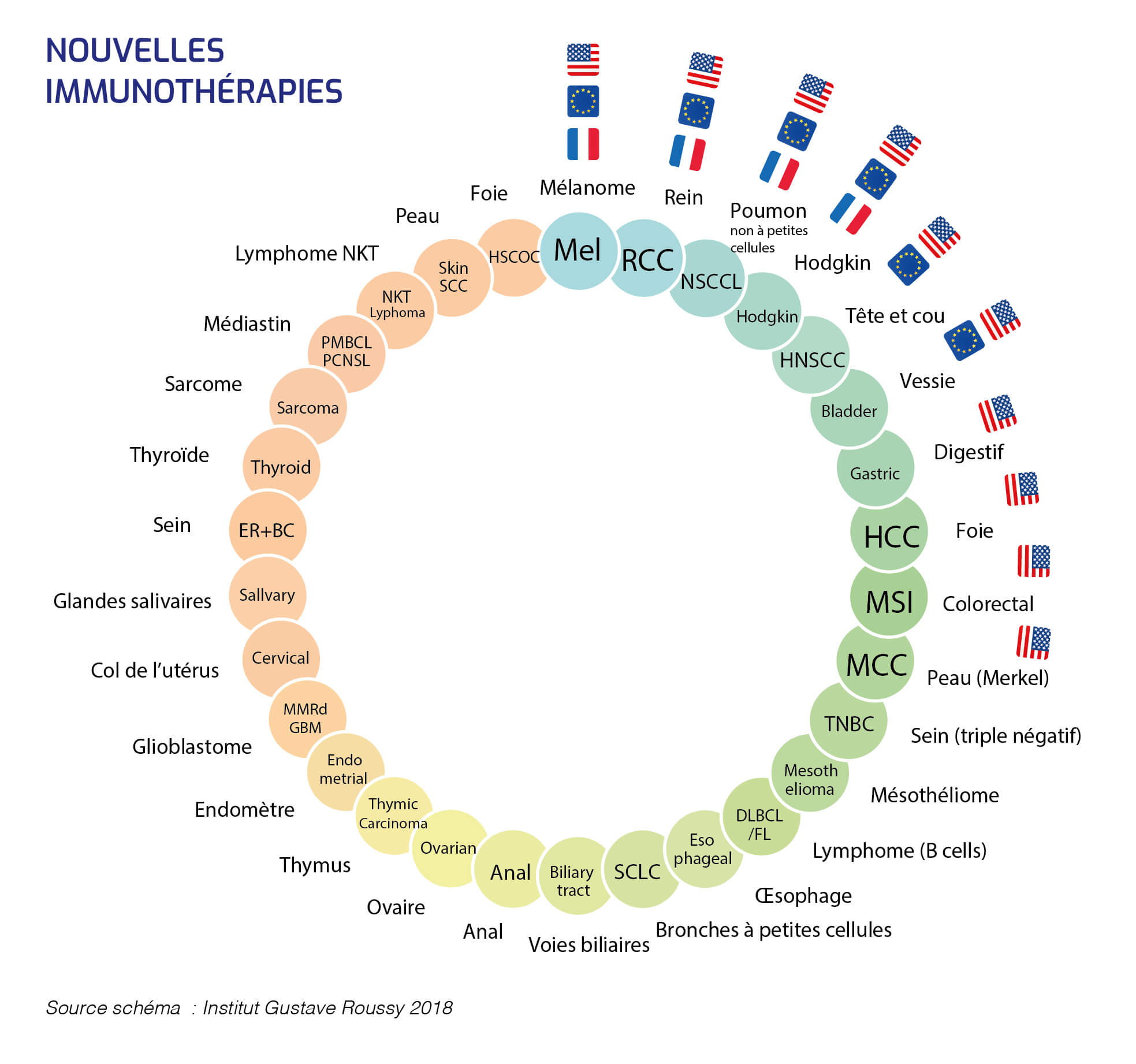
Le retard français
Après l'obtention de l'autorisation de mise sur le marché (AMM) - au niveau européen pour les produits innovants -, la commercialisation des médicaments suit en France une procédure séquencée en vue de leur admission au remboursement.
Les produits sont d'abord évalués par la Haute Autorité de santé (HAS), puis par le CEPS où leur prix est fixé dans un cadre conventionnel.
En France, le délai entre l'obtention d'une AMM et l'arrivée d'un nouveau médicament s'élève en moyenne à 500 jours, contre 100 jours en Allemagne, au-delà des 180 jours prescrits par la directive européenne en vigueur.
L'oncologie aggrave cette tendance (656 jours pour les anticancéreux délivrés en ville, 543 jours pour les anticancéreux de la liste en sus ).
Il convient de remarquer que cette situation n'intègre pas le dispositif des ATU (autorisations temporaires d'utilisation), ce qui minore quelque peu les délais beaucoup trop longs.
Les ATU : une solution pertinente pour un accès précoce aux traitements innovants, mais qui ne peut être généralisée
La France a construit un système de recherche clinique et d'accès précoce aux avancées thérapeutiques avec les autorisations temporaires d'utilisation.
Il offre la possibilité aux patients de bénéficier de traitements en cours de développement.
Quasiment unique en Europe, le dispositif des ATU a permis, depuis 1994, de mettre un grand nombre de médicaments innovants à la disposition des malades atteints de pathologies graves ou rares et en situation d'impasse thérapeutique, parfois plus d'un an avant la délivrance de l'AMM.
Avec l'arrivée de traitements de précision très innovants, le coût des ATU a fortement augmenté, atteignant 1 milliard d'euros en 2016, contre 110 millions d'euros en 2013 - 80 % de la charge étant représentée par quatre traitements anticancéreux.
Le risque est que ce système, conçu comme temporaire et dérogatoire, se développe considérablement en tant que dispositif structuré d'accès précoce au marché pour un nombre important de patients, au risque de déséquilibrer les logiques d'accès.
Les médicaments ayant bénéficié d'une ATU ont été disponibles pour les patients 210 jours avant l'AMM (c'est-à-dire 318 jours avant l'Allemagne et le Royaume-Uni), mais ils ne l'ont été pour seulement 10 % de la population concernée.
Autrement dit, l'ATU est loin de permettre d'atteindre l'intégralité de la population éligible aux innovations thérapeutiques.
Par ailleurs, les lois successives de financement de la Sécurité sociale ont modifié profondément ce régime le rendant très compliqué, voire inopérant.
Les enjeux relatifs au financement des actes de biologie innovants
Les tests dits "compagnons" sont indispensables au développement de la médecine de précision en oncologie.
L'enjeu est majeur en termes de pertinence des soins et pour la qualité de vie des patients, en permettant les adaptations thérapeutiques.
Toutefois, le mode de prise en charge de ces tests est un frein à leur développement : l'enveloppe annuelle de 380 millions d'euros (en 2017) ne permet pas de financer l'ensemble des actes réalisés par les établissements de santé, d'où des pratiques inégales.
Le sentiment des différentes parties prenantes, même si leurs préoccupations et solutions sont différentes, est celui d'un système qui subit l'innovation et son coût, et dont les procédures standardisées sont devenues inadaptées au contexte actuel de l'innovation.
Un scénario inquiétant se profile pour certains : les patients informés des traitements innovants difficilement accessibles engageront des dépenses à l'étranger, certains n'y accéderont pas, soit pour des raisons d'informations, ou parce que l'hôpital de leur région n'est pas en situation de les délivrer (cas de l'Avastin® pris en charge dans les CHU de Lyon, mais pas dans les hôpitaux de taille moyenne, ou du Dacogen® pris en charge à Paris et dans l'Ouest, mais peu ailleurs...).
Les débats de société sur l'accès aux soins conduisent à des tensions entre citoyens.
(Chantier 3 "Transformer les mécanismes d'évaluation et gagner en efficacité administrative pour permettre aux patients d'accéder plus vite aux traitements innovants").
Extrait de Santé 2030 - Partie 4 : 10 chantiers pour construire 2030. Retrouvez l'intégralité de l'étude sur le site.